Note
Go to the end to download the full example code.
Running Molecular Dynamics with ASE¶
This tutorial shows how to use metatensor atomistic models to run Molecular Dynamics (MD) simulation using ASE. ASE is not the only way to run MD with metatensor models, but it is very easy to setup and use.
from typing import Dict, List, Optional
# tools to run the simulation
import ase.build
import ase.md
import ase.md.velocitydistribution
import ase.units
# tools for visualization
import ase.visualize.plot
import chemiscope
import matplotlib.pyplot as plt
# the usuals suspects
import numpy as np
import torch
from metatensor.torch import Labels, TensorBlock, TensorMap
from metatensor.torch.atomistic import (
MetatensorAtomisticModel,
ModelCapabilities,
ModelMetadata,
ModelOutput,
System,
)
# Integration with ASE calculator for metatensor atomistic models
from metatensor.torch.atomistic.ase_calculator import MetatensorCalculator
The energy model¶
To run simulations, we’ll need a model. For simplicity and keeping this tutorial short, we’ll use a model for Einstein’s solid, where all atoms oscillate around their equilibrium position as harmonic oscillators, but don’t interact with one another. The energy of each atoms is given by:
where \(k\) is a harmonic force constant, \(\vec{r_i}\) the position of atom \(i\) and \(\vec{r_i}^0\) the position of atom \(i\) at equilibrium. The energy of the whole system is then
Let’s implement this model! You can check the Exporting a model tutorial for more information on how to define your own custom models.
class HarmonicModel(torch.nn.Module):
def __init__(self, force_constant: float, equilibrium_positions: torch.Tensor):
"""Create an ``HarmonicModel``.
:param force_constant: force constant, in ``energy unit / (length unit)^2``
:param equilibrium_positions: torch tensor with shape ``n x 3``, containing the
equilibrium positions of all atoms
"""
super().__init__()
assert force_constant > 0
self.force_constant = force_constant
self.equilibrium_positions = equilibrium_positions
def forward(
self,
systems: List[System],
outputs: Dict[str, ModelOutput],
selected_atoms: Optional[Labels],
) -> Dict[str, TensorMap]:
if list(outputs.keys()) != ["energy"]:
raise ValueError(
"this model can only compute 'energy', but `outputs` contains other "
f"keys: {', '.join(outputs.keys())}"
)
# we don't want to worry about selected_atoms yet
if selected_atoms is not None:
raise NotImplementedError("selected_atoms is not implemented")
if outputs["energy"].per_atom:
raise NotImplementedError("per atom energy is not implemented")
# compute the energy for each system by adding together the energy for each atom
energy = torch.zeros((len(systems), 1), dtype=systems[0].positions.dtype)
for i, system in enumerate(systems):
assert len(system) == self.equilibrium_positions.shape[0]
r0 = self.equilibrium_positions
energy[i] += torch.sum(self.force_constant * (system.positions - r0) ** 2)
# add metadata to the output
block = TensorBlock(
values=energy,
samples=Labels("system", torch.arange(len(systems)).reshape(-1, 1)),
components=[],
properties=Labels("energy", torch.tensor([[0]])),
)
return {
"energy": TensorMap(keys=Labels("_", torch.tensor([[0]])), blocks=[block])
}
Initial simulation state¶
Now that we have a model for the energy of our system, let’s create some initial
simulation state. We’ll build a 3x3x3 super cell of diamond carbon. In practice, you
could also read the initial state from a file using ase.io.read().
primitive = ase.build.bulk(name="C", crystalstructure="diamond", a=3.567)
ax = ase.visualize.plot.plot_atoms(primitive, radii=0.5)
ax.set_xlabel("$\\AA$")
ax.set_ylabel("$\\AA$")
plt.show()
And now let’s make a super cell with this
atoms = ase.build.make_supercell(primitive, 3 * np.eye(3))
ax = ase.visualize.plot.plot_atoms(atoms, radii=0.5)
ax.set_xlabel("$\\AA$")
ax.set_ylabel("$\\AA$")
plt.show()
We’ll also need to initialize the velocities of the atoms to match our simulation temperature.
# The atoms start with zero velocities, hence zero kinetic energy and zero temperature
print("before:")
print("kinetic energy =", atoms.get_kinetic_energy(), "eV")
print("temperature =", atoms.get_temperature(), "K")
ase.md.velocitydistribution.MaxwellBoltzmannDistribution(atoms, temperature_K=300)
print("\nafter:")
print("kinetic energy =", atoms.get_kinetic_energy(), "eV")
print("temperature =", atoms.get_temperature(), "K")
before:
kinetic energy = 0.0 eV
temperature = 0.0 K
after:
kinetic energy = 2.1787522298536093 eV
temperature = 312.14047303066786 K
Running the simulation¶
The final step to run the simulation is to register our model as the energy calculator
for these atoms. This is the job of
ase_calculator.MetatensorCalculator, which takes either an instance of
MetatensorAtomisticModel or the path to a pre-exported model, and allow to
use it to compute the energy, forces and stress acting on a system.
# define & wrap the model, using the initial positions as equilibrium positions
model = HarmonicModel(
force_constant=3.14159265358979323846,
equilibrium_positions=torch.tensor(atoms.positions),
)
capabilities = ModelCapabilities(
outputs={
"energy": ModelOutput(quantity="energy", unit="eV", per_atom=False),
},
atomic_types=[6],
interaction_range=0.0,
length_unit="Angstrom",
supported_devices=["cpu"],
dtype="float32",
)
# we don't want to bother with model metadata, so we define it as empty
metadata = ModelMetadata()
wrapper = MetatensorAtomisticModel(model.eval(), metadata, capabilities)
# Use the wrapped model as the calculator for these atoms
atoms.calc = MetatensorCalculator(wrapper)
We’ll run the simulation in the constant volume/temperature thermodynamics ensemble
(NVT or Canonical ensemble), using a Langevin thermostat for time integration. Please
refer to the corresponding documentation (ase.md.langevin.Langevin) for
more information!
integrator = ase.md.Langevin(
atoms,
timestep=1.0 * ase.units.fs,
temperature_K=300,
friction=0.1 / ase.units.fs,
)
# collect some data during the simulation
trajectory = []
potential_energy = []
kinetic_energy = []
total_energy = []
temperature = []
for step in range(800):
# run a single simulation step
integrator.run(1)
# collect data about the simulation
potential_energy.append(atoms.get_potential_energy())
kinetic_energy.append(atoms.get_kinetic_energy())
total_energy.append(atoms.get_total_energy())
temperature.append(atoms.get_temperature())
if step % 10 == 0:
trajectory.append(atoms.copy())
We can use chemiscope to visualize the trajectory
viewer_settings = {"bonds": False, "playbackDelay": 70}
chemiscope.show(trajectory, mode="structure", settings={"structure": [viewer_settings]})

We can also look at the time evolution of some physical constants for our system:
fig, ax = plt.subplots(1, 2, figsize=(8, 5))
ax[0].plot(range(800), potential_energy, label="potential energy")
ax[0].plot(range(800), kinetic_energy, label="kinetic energy")
ax[0].plot(range(800), total_energy, label="total energy")
ax[0].legend()
ax[0].set_xlabel("t / fs")
ax[0].set_ylabel("energy / eV")
ax[1].plot(range(800), temperature)
ax[1].set_xlabel("t / fs")
ax[1].set_ylabel("temperature / K")
fig.tight_layout()
fig.show()
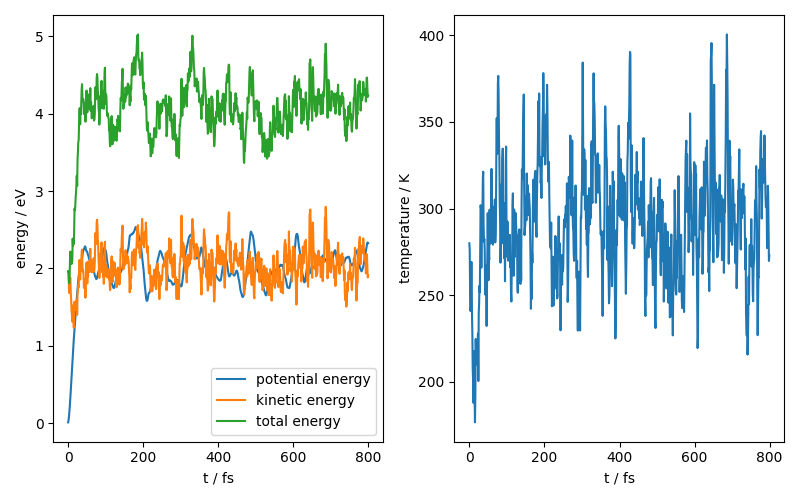
Using a pre-exported model¶
As already mentioned, ase_calculator.MetatensorCalculator also work with a
pre-exported model, meaning you can also run simulations without defining or
re-training a model:
wrapper.save("exported-model.pt")
atoms.calc = MetatensorCalculator("exported-model.pt")
print(atoms.get_potential_energy())
integrator.run(10)
1.9903477430343628
True
Total running time of the script: (0 minutes 2.452 seconds)